Pangenomes reveal differences between cattle and their wild relatives
- D-USYS
- Institute of Agricultural Sciences
- Research
- Agricultural sciences
A collaboration between the Animal Genomics group at ETH Zurich and the U.S. Meat Animal Research Center (USDA-ARS) investigated which methods of sequencing and assembling produce high-quality bovine genomes, how pangenomes robustly combine such assemblies, and how these new resources reveal hitherto inaccessible trait-associated DNA variants.
Consortia-scale genome projects tend to recommend specific protocols for sequencing and assembly to maximize quality and consistency across contributors. However, since sequencing and assembling technologies evolve constantly and rapidly, these recommendations quickly become outdated.
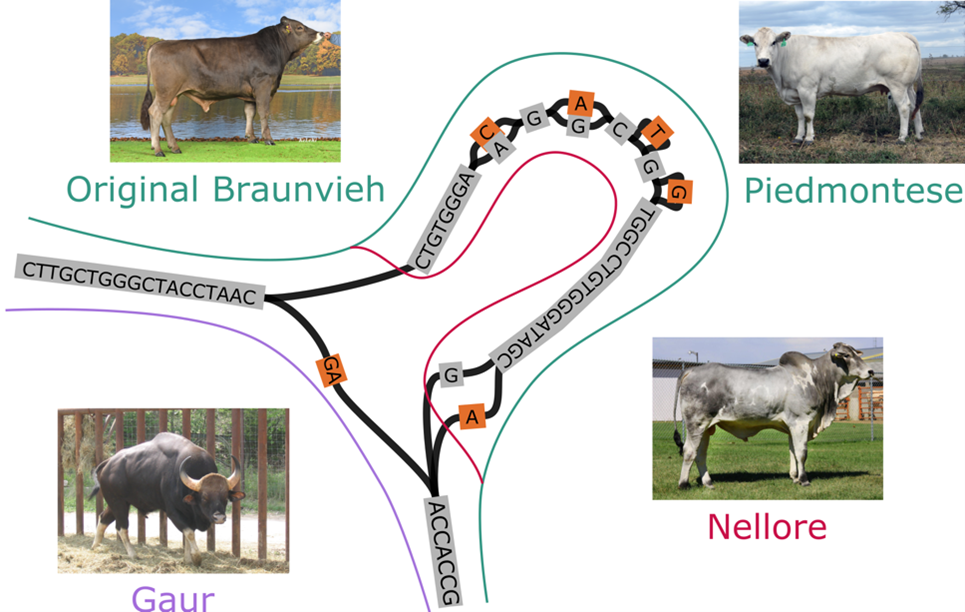
A comprehensive evaluation of current technologies revealed the best approaches to sequencing and assembly for three bovine crosses that varied greatly in heterozygosity. While the genome assemblies differed slightly, the resulting structural variant-based pangenomes were broadly consistent regardless of the protocol used to generate the input assemblies. The pangenomes revealed larges structural differences between individual genomes suggesting that pangenome-wide association testing can detect trait-associated variants. This approach is particularly promising for types of variants that were challenging to resolve so far with linear reference-based methods.
Taken together, these results suggest that large-scale projects should welcome as many contributions as possible to compile comprehensive non-linear references, rather than requiring rigid protocols that likely will exclude participation from groups with different financial and logistical constraints. This is especially important for large international efforts like the Bovine Pangenome Consortium that aims to build a community-accepted pangenome from several hundred breeds of global cattle and their wild relatives.
Reference
Leonard AS, Crysnanto D, Fang ZH et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. Nat Commun 13, 3012 (2022). doi: external page 10.1038/s41467-022-30680-2.